

理化学研究所
静岡県立総合病院
静岡県立大学
静岡県立総合病院
静岡県立大学
ヒトの特徴を形作る遺伝多型
-非ヨーロッパ系集団における最大規模のゲノム研究で明らかに-
概要
理化学研究所(理研)生命医科学研究センターゲノム解析応用研究チームの寺尾知可史チームリーダー(静岡県立総合病院臨床研究部免疫研究部長、静岡県立大学薬学部ゲノム病態解析分野特任教授)らの国際共同研究グループは、日本人におけるヒトの特徴(量的形質[1])に関係する遺伝多型[2]を多数明らかにしました。
本研究の成果は、ヒトの量的形質に影響を与える遺伝子や分子の理解を深め、個別化医療におけるゲノム情報の応用に貢献することが期待されます。
国際共同研究グループは、大規模な日本人の遺伝多型タイピングデータを用いて、日本人の全ゲノムシークエンス[3]データを基に、高精度な遺伝多型の推定を行い、臨床情報との関連を調査しました。また、量的形質の原因遺伝多型である可能性が高い遺伝多型を統計学的に決定しました。その結果、身長、体重、血圧、コレステロール値といった身体的・医学的な量的形質に影響を与える数多くの遺伝多型のリストを作成しました。
例えば、IL6遺伝子の3’非翻訳領域に存在する日本人特有のまれな遺伝多型が、免疫機能を変化させ、結核の罹患(りかん)リスクに影響を与えることが判明しました。この遺伝多型は、日本人を含む東アジア人集団に特有であり、他の集団では発見が難しく、日本人におけるゲノム研究の重要性を示しています。
本研究は、非ヨーロッパ系集団において最大規模であり、東アジア人集団における遺伝多型とヒトの特徴との関係を解明するための重要なリソースとなることが期待されます。
本研究は、科学雑誌『Nature Genetics』オンライン版(10月3日付:日本時間10月3日)に掲載されました。
本研究の成果は、ヒトの量的形質に影響を与える遺伝子や分子の理解を深め、個別化医療におけるゲノム情報の応用に貢献することが期待されます。
国際共同研究グループは、大規模な日本人の遺伝多型タイピングデータを用いて、日本人の全ゲノムシークエンス[3]データを基に、高精度な遺伝多型の推定を行い、臨床情報との関連を調査しました。また、量的形質の原因遺伝多型である可能性が高い遺伝多型を統計学的に決定しました。その結果、身長、体重、血圧、コレステロール値といった身体的・医学的な量的形質に影響を与える数多くの遺伝多型のリストを作成しました。
例えば、IL6遺伝子の3’非翻訳領域に存在する日本人特有のまれな遺伝多型が、免疫機能を変化させ、結核の罹患(りかん)リスクに影響を与えることが判明しました。この遺伝多型は、日本人を含む東アジア人集団に特有であり、他の集団では発見が難しく、日本人におけるゲノム研究の重要性を示しています。
本研究は、非ヨーロッパ系集団において最大規模であり、東アジア人集団における遺伝多型とヒトの特徴との関係を解明するための重要なリソースとなることが期待されます。
本研究は、科学雑誌『Nature Genetics』オンライン版(10月3日付:日本時間10月3日)に掲載されました。
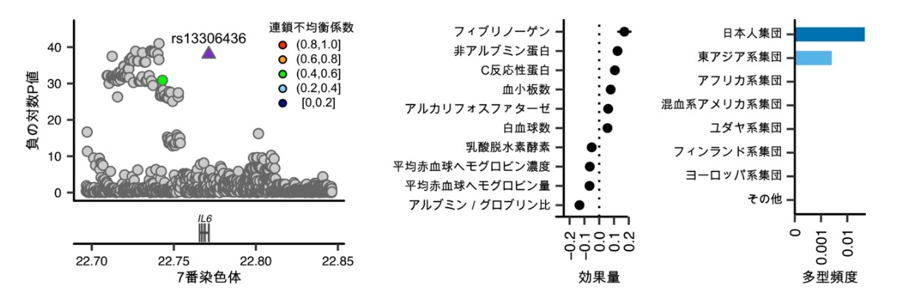
IL6遺伝子の3’非翻訳領域に存在し免疫機能に影響を与える日本人特有のまれな遺伝多型
背景
私たちのゲノム上に存在する遺伝多型は、遺伝子の機能やその発現変化を通じて、私たちの身体的・医学的特徴に影響を与えます。ゲノムワイド関連解析(GWAS)[4]は、私たちが持って生まれた遺伝多型と個人のさまざまな量的形質を結び付けることで、その量的形質に関与する遺伝子を特定するための研究手法です。これまでに、日本人を含む多くの集団でGWASが実施され、数千から数万の遺伝子と病気やヒトの量的形質の関連が報告されてきました。
GWASは遺伝子とヒトの量的形質をひも付ける強力な手法ですが、従来の遺伝多型の同定方法やデータ分析手法では、関連遺伝シグナルの存在する領域が分かっても、実際の原因遺伝子や原因遺伝多型の特定が難しい場合がありました。そこで本研究では、次の三つのアプローチを採用しました。(1)日本人の遺伝背景に合致した遺伝的インピュテーション[5]のための参照データを作成し、遺伝多型の同定精度を高めること、(2)複数のバイオバンクデータを統合解析することで統計学的検出力を向上させること、(3)統計学的ファインマッピング[6]を応用し、原因遺伝子や原因遺伝多型の同定を行うことです。
GWASは遺伝子とヒトの量的形質をひも付ける強力な手法ですが、従来の遺伝多型の同定方法やデータ分析手法では、関連遺伝シグナルの存在する領域が分かっても、実際の原因遺伝子や原因遺伝多型の特定が難しい場合がありました。そこで本研究では、次の三つのアプローチを採用しました。(1)日本人の遺伝背景に合致した遺伝的インピュテーション[5]のための参照データを作成し、遺伝多型の同定精度を高めること、(2)複数のバイオバンクデータを統合解析することで統計学的検出力を向上させること、(3)統計学的ファインマッピング[6]を応用し、原因遺伝子や原因遺伝多型の同定を行うことです。
研究手法と成果
現在のGWASでは、遺伝多型の検出精度を高めるために、参照データ(リファレンスパネル[7])を用いた遺伝的インピュテーションという手法が用いられます。しかし、これまでに用いられてきた参照データの多くはヨーロッパ系集団から作成されたものであり、日本人のゲノム情報に適合するデータは十分ではありませんでした。国際共同研究グループは、日本人集団のゲノム情報の補完精度を向上させるために、日本人の全ゲノムシークエンスデータを基に参照データを作成しました。これにより、被験者のゲノム情報を高精度に補完することが可能となりました。
この高精度なゲノム情報を用いて、日本人集団の量的形質と遺伝多型との関係を解析しました。その結果、検討した63の日本人集団の量的形質とゲノム上の4,423の領域との関連を明らかにしました。その中で、601の領域はこれまでのGWASでは発見されていなかったものでした。
例えば、トロポニン遺伝子と心臓の機能との関連性が発見されました(図1)。トロポニン遺伝子は心臓の重要な構成タンパク質を産生し、拡張型心筋症を引き起こす原因遺伝子として知られていますが、これまでの心不全を対象としたGWASではその関連は検出されていませんでした。今回の研究では、トロポニン遺伝子の構造を変化させる遺伝多型と心臓の機能との強い関連が示され、この遺伝多型を持つ人は心臓の機能が低下し、心不全に罹患するリスクが4.5倍高いことが明らかになりました。
この高精度なゲノム情報を用いて、日本人集団の量的形質と遺伝多型との関係を解析しました。その結果、検討した63の日本人集団の量的形質とゲノム上の4,423の領域との関連を明らかにしました。その中で、601の領域はこれまでのGWASでは発見されていなかったものでした。
例えば、トロポニン遺伝子と心臓の機能との関連性が発見されました(図1)。トロポニン遺伝子は心臓の重要な構成タンパク質を産生し、拡張型心筋症を引き起こす原因遺伝子として知られていますが、これまでの心不全を対象としたGWASではその関連は検出されていませんでした。今回の研究では、トロポニン遺伝子の構造を変化させる遺伝多型と心臓の機能との強い関連が示され、この遺伝多型を持つ人は心臓の機能が低下し、心不全に罹患するリスクが4.5倍高いことが明らかになりました。
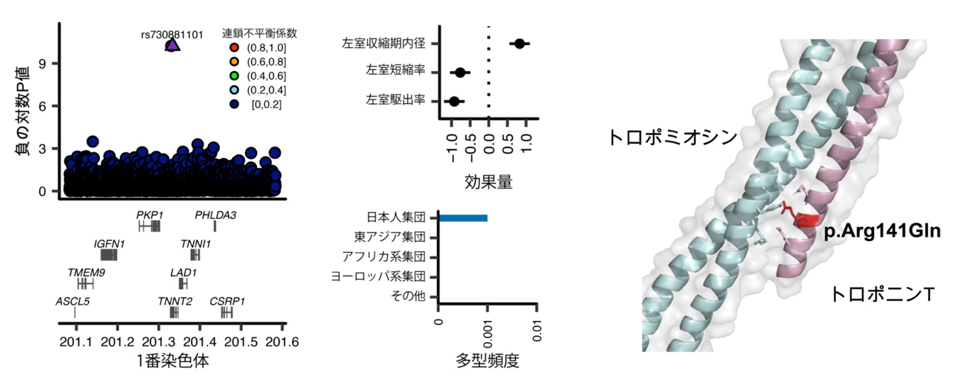
図1 心機能低下・心不全を来す日本人特有のTNNT2遺伝多型
左)トロポニン遺伝子の構造を変化させる遺伝多型(rs730881101)がTNNT2遺伝子に見つかった。負の対数P値:それぞれの変異の統計的有意性を表すP値の底を10とした対数を取り、符号を転換したもの。0.01の場合、対数変換によって「-2」となり、符号の転換によって「2」となる。非常に小さなP値を表示するのに使用する。連鎖不平衡係数:変異同士の連鎖(相関)を表す。係数が高いほど連鎖(相関が高い)と解釈できる。図では最も統計学的に有意であった変異(紫の三角で示されている)に対する連鎖不平衡係数が各点の色として表されており、rs730881101以外のほとんどが0-0.2の範囲([0,0.2])の黒丸になっている。
中上)この遺伝多型は心臓の機能を低下させ、心不全のリスクを高める。効果量:変異当たりの形質に与える影響を示す。本研究では効果量の単位は形質の一標準偏差に統一されている。
中下)この遺伝多型は日本人集団に多い。
右)今回見つかった遺伝子変異(p.Arg141Gln)はトロポニンTとトロポミオシンの結合部位に存在する。
中上)この遺伝多型は心臓の機能を低下させ、心不全のリスクを高める。効果量:変異当たりの形質に与える影響を示す。本研究では効果量の単位は形質の一標準偏差に統一されている。
中下)この遺伝多型は日本人集団に多い。
右)今回見つかった遺伝子変異(p.Arg141Gln)はトロポニンTとトロポミオシンの結合部位に存在する。
さらに、非コーディング多型[8](遺伝子の機能に直接関わらない遺伝多型)についても、強い影響を持つ遺伝多型を発見しました。例えば、FLT3遺伝子のイントロンと呼ばれる領域に存在する遺伝多型です。イントロンに存在する遺伝多型は一般的には遺伝子の機能に大きく関与しないと考えられますが、この遺伝多型は、近年開発された機械学習を用いた遺伝多型機能解析アルゴリズム(Splice-AI)によって、強い遺伝子破壊効果があることが示されました。実際に遺伝多型を導入したベクター(運び屋)を用いて調査した結果、この遺伝多型は正常な遺伝子スプライシング[9]を強く阻害し(図2)、FLT3の遺伝子機能が破壊されると、関節リウマチや全身性エリテマトーデスなどの膠原(こうげん)病の罹患リスクが上昇することも明らかになりました。この遺伝多型は日本人以外にも東アジア人集団全体に見られましたが、他の集団では観察されず、東アジア人集団に特有の遺伝多型であることが示されました。
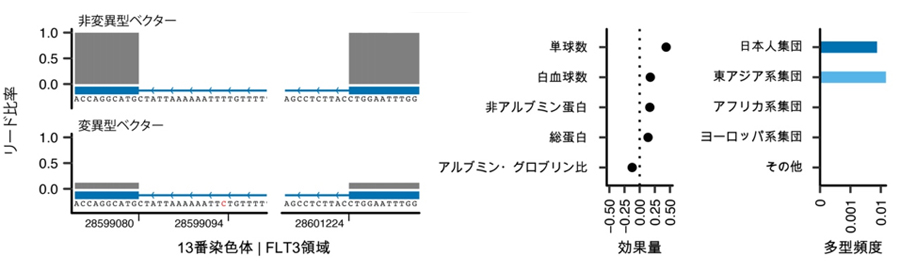
図2 スプライシング異常を介してFLT3遺伝子機能を破壊するイントロン遺伝多型
左)変異型と非変異型のFLT3遺伝子を導入したベクター(運び屋)を用いて遺伝子の発現パターンを観察した。変異型ベクターではFLT3遺伝子の発現低下が確認された。遺伝子スプライシングが強く阻害されたと見られる。
中)FLT3遺伝子における遺伝多型は白血球数をはじめとした免疫的形質に影響を与える。
右)FLT3遺伝子の機能を破壊する遺伝多型は、アフリカ系やヨーロッパ系の集団などには見られず、日本人集団、東アジア人集団に多く観察される。
中)FLT3遺伝子における遺伝多型は白血球数をはじめとした免疫的形質に影響を与える。
右)FLT3遺伝子の機能を破壊する遺伝多型は、アフリカ系やヨーロッパ系の集団などには見られず、日本人集団、東アジア人集団に多く観察される。
これらの結果から、精密な遺伝多型の同定手法と大規模な被験者数でGWASを実施すると、遺伝子の機能や活性に関わる遺伝多型を同定できることが期待されました。この期待を裏付けるように、量的形質と強い関連が見られた遺伝多型は、コーディング多型を中心とした遺伝子の機能的コンポーネントに集積していることが分かりました。また、英国バイオバンクのヨーロッパ系集団のデータと比較したところ、この集積は日本人集団とヨーロッパ系集団に共通していることが示されました(図3左)。特に、遺伝子前後に付随する5’非翻訳領域と3’非翻訳領域に強い集積が見られました。また、今回発見された統計学的に原因遺伝多型である可能性の高いものは、より東アジア人集団に特異的なものが多いことが分かりました(図3右)。
これらのことは、ヒトの量的形質を規定する遺伝多型の性質やその背景にあるメカニズムは集団間で共通であるものの、その遺伝多型自体は集団間で異なり、さまざまな人類集団の遺伝データを解析することの重要性を示唆しています。
これらのことは、ヒトの量的形質を規定する遺伝多型の性質やその背景にあるメカニズムは集団間で共通であるものの、その遺伝多型自体は集団間で異なり、さまざまな人類集団の遺伝データを解析することの重要性を示唆しています。
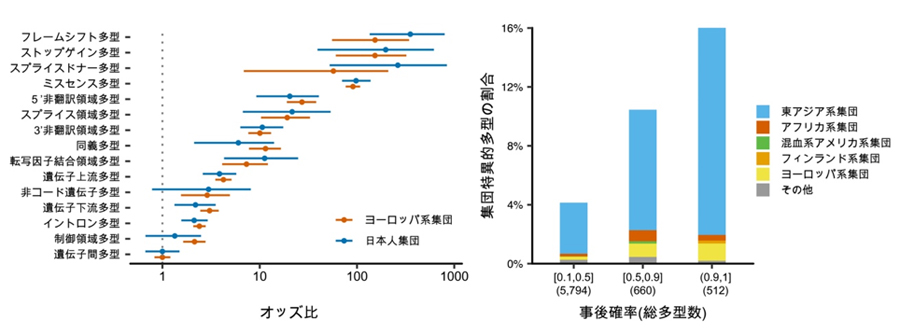
図3 集団間で共通する遺伝的構造と集団特異的遺伝多型の存在
左)日本人集団とヨーロッパ系集団の遺伝多型の集積を比較した。この集積は両者に共通している。中でも遺伝子前後に付随する5’非翻訳領域と3’非翻訳領域に強い集積が見られた。オッズ比:原因遺伝多型の当該遺伝子領域への集積を表す。この図では事後確率90%以上の原因遺伝多型が遺伝多型に対してどの程度集積しているかを示している。
右)原因遺伝多型における集団特異的遺伝多型の割合。全ての原因遺伝多型のうち、gnomADデータベースにおける六つの参照集団において特異的に見つかる遺伝多型の割合を示している。原因遺伝多型である確率(事後確率)が高くなるほど東アジア人集団特異的な遺伝多型の割合が増えることがわかる。
右)原因遺伝多型における集団特異的遺伝多型の割合。全ての原因遺伝多型のうち、gnomADデータベースにおける六つの参照集団において特異的に見つかる遺伝多型の割合を示している。原因遺伝多型である確率(事後確率)が高くなるほど東アジア人集団特異的な遺伝多型の割合が増えることがわかる。
3’非翻訳領域は、遺伝子の発現量を調整する上で重要な部分と考えられています。今回、IL6(インターロイキン6)[10]をコードするIL6遺伝子の、3’非翻訳領域に存在する遺伝多型とヒトの量的形質との関係が発見されました。この遺伝多型は、フィブリノーゲンやC反応性タンパクの上昇といった炎症に関連する検査項目の変化と強く関連していました(図4)。さらに、この遺伝多型を持つ人は結核に罹患しにくいことも明らかになりました。IL6の量を調整するRegnase-1[11]というタンパク質の働きが、この遺伝多型によって阻害され、結果的にIL6の量が増加することで、炎症反応が強まり結核に対する感染防御として機能している可能性があります。
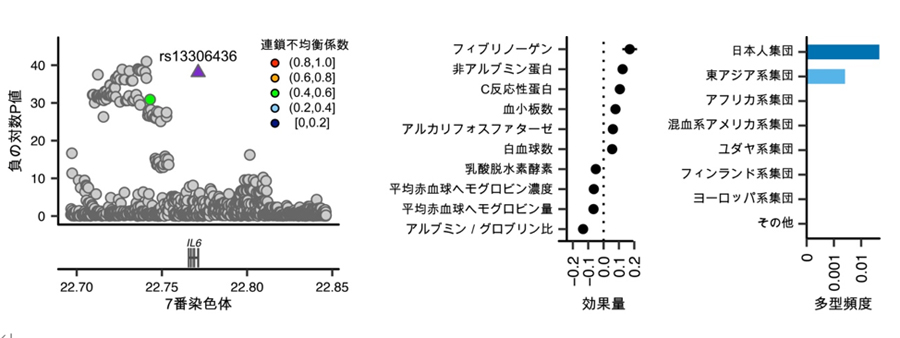
図4 炎症の誘導によって結核感染耐性を示すIL6遺伝多型
左)IL6遺伝子の3’非翻訳領域にある遺伝多型「rs13306436」にヒトの量的形質との関係が見つかった。
中)この遺伝多型はフィブリノーゲンやC反応性タンパクなどの上昇といった炎症関連の検査項目の変化と関連していた。
右)この遺伝多型の頻度は日本人集団や東アジア人集団で多くなっていることが分かった。
中)この遺伝多型はフィブリノーゲンやC反応性タンパクなどの上昇といった炎症関連の検査項目の変化と関連していた。
右)この遺伝多型の頻度は日本人集団や東アジア人集団で多くなっていることが分かった。
今後の期待
ヒトの量的形質と関連付けられた遺伝子および遺伝多型のリストは、今後の創薬や精密医療の基礎データとして重要な役割を果たすことが期待されます。
また、今回の研究結果は、遺伝データおよび解析手法の進歩により、GWASが原因遺伝子や遺伝多型の検出手法としても有用であることを裏付けています。今後、研究対象とする集団やヒトの量的形質を増やすことで、さらに多くの発見が期待されます。
今回の研究で得られたデータは、理研のウェブサイトJenger(http://jenger.riken.jp/en/)およびNBDC(https://biosciencedbc.jp/en/)を通じて公開される予定であり、さまざまな研究者による再解析や新たな発見の促進が期待されます。
また、今回の研究結果は、遺伝データおよび解析手法の進歩により、GWASが原因遺伝子や遺伝多型の検出手法としても有用であることを裏付けています。今後、研究対象とする集団やヒトの量的形質を増やすことで、さらに多くの発見が期待されます。
今回の研究で得られたデータは、理研のウェブサイトJenger(http://jenger.riken.jp/en/)およびNBDC(https://biosciencedbc.jp/en/)を通じて公開される予定であり、さまざまな研究者による再解析や新たな発見の促進が期待されます。
論文情報
<タイトル>
Population-specific putative causal variants shape quantitative traits
<著者名>
Satoshi Koyama, Xiaoxi Liu, Yoshinao Koike, Keiko Hikino, Masaru Koido, Wei Li, Kotaro Akaki, Kohei Tomizuka, Shuji Ito, Nao Otomo, Hiroyuki Suetsugu, Soichiro Yoshino, Masato Akiyama, Kohei Saito, Yuki Ishikawa, Christian Benner, Pradeep Natarajan, Patrick T. Ellinor, Taisei Mushiroda, Momoko Horikoshi, Masashi Ikeda, Nakao Iwata, Koichi Matsuda, Biobank Japan Project, Shumpei Niida, Kouichi Ozaki, Yukihide Momozawa, Shiro Ikegawa, Osamu Takeuchi, Kaoru Ito, Chikashi Terao
<雑誌>
Nature Genetics
<DOI>
10.1038/s41588-024-01913-5
Population-specific putative causal variants shape quantitative traits
<著者名>
Satoshi Koyama, Xiaoxi Liu, Yoshinao Koike, Keiko Hikino, Masaru Koido, Wei Li, Kotaro Akaki, Kohei Tomizuka, Shuji Ito, Nao Otomo, Hiroyuki Suetsugu, Soichiro Yoshino, Masato Akiyama, Kohei Saito, Yuki Ishikawa, Christian Benner, Pradeep Natarajan, Patrick T. Ellinor, Taisei Mushiroda, Momoko Horikoshi, Masashi Ikeda, Nakao Iwata, Koichi Matsuda, Biobank Japan Project, Shumpei Niida, Kouichi Ozaki, Yukihide Momozawa, Shiro Ikegawa, Osamu Takeuchi, Kaoru Ito, Chikashi Terao
<雑誌>
Nature Genetics
<DOI>
10.1038/s41588-024-01913-5
補足説明
[1] 量的形質
形質とは個人の持つ性質のことをいい、病気の有無や身長などが含まれる。この中でも特に身長や白血球の数などは連続値であるため、量的形質と呼ばれる。本研究ではこの量的形質に着目して研究を行った。
[2] 遺伝多型
ゲノムは個人間でわずかな違いがある。この違いは一塩基の置換(一塩基多型)から、数十塩基までの挿入や欠失(挿入多型、欠失多型)、さらに大きな挿入や欠失(構造多型)などを含む。これらの遺伝多型の有無とヒトの形質を結び付けるのがGWAS([4]参照)である。
[3] 全ゲノムシークエンス
ヒト個体の全ての遺伝情報(ゲノム)を網羅的に解読する技術。ゲノムは約30億対のDNA塩基対から構成されており、この全体を読み取ることで、個体の遺伝的構成を詳細に把握できる。全ゲノムシークエンスはリファレンスパネル([7]参照)の作成に重要な役割を担う。
[4] ゲノムワイド関連解析(GWAS)
ヒトの形質(例:身長、病気のリスクなど)を目的変数とし、数百万に及ぶ遺伝多型を説明変数として統計学的検定を行い、ヒトの形質と有意に関連する遺伝多型を特定する手法。GWASはGenome-Wide Association Studyの略。
[5] 遺伝的インピュテーション
ゲノム解析において実際に測定されていない遺伝多型の情報を補完する手法。既知の遺伝情報(参照データ)を基に、欠損している部分の遺伝多型を推定し、データセットに含まれる遺伝多型数を増加させることで解析の精度を高める。
[6] 統計学的ファインマッピング
GWASで特定された関連領域の中から、原因となる可能性の高い遺伝多型を絞り込むための解析手法。GWASでは、広範なゲノム領域が検出されるが、その中には多くの遺伝多型が含まれている。統計学的ファインマッピングでは研究集団の連鎖不均衡(Linkage Disequilibrium:LD)情報を活用し、これらの遺伝多型の中から原因遺伝多型を特定する精度を向上させる。従来は計算量が多く、10万人を超える被験者や多数の遺伝領域に対しては実施が困難であったが、高効率なアルゴリズムの開発により可能となっている。
[7] リファレンスパネル
遺伝的インピュテーションにおける基準データであり、既知の遺伝多型情報を集めたデータセットのこと。全ゲノムシークエンスに基づいて収集された日本人集団の遺伝情報が多く含まれており、これを使用することで、実際に測定されていない遺伝多型を高精度で推定することができる。特に、対象集団の遺伝的背景と一致するリファレンスパネルを用いることで、インピュテーションの精度が向上する。
[8] 非コーディング多型
DNA配列の中でタンパク質の構造に直接関与しない領域に生じる遺伝多型。これらの非コーディング領域には、プロモーターやエンハンサー、イントロン、5’非翻訳領域、3’非翻訳領域などが含まれる。非コーディング多型は、タンパク質の構造には直接影響を与えないが、遺伝子の発現レベル、スプライシングの効率、転写因子の結合、mRNAの安定性や翻訳効率に影響を与え、結果として間接的に細胞の機能やタンパク質の産生に影響を与える可能性がある。
[9] スプライシング
前駆体mRNA(pre-mRNA)から不要な部分(イントロン)を除去し、必要な部分(エクソン)をつなぎ合わせて成熟したmRNAを形成する過程。このスプライシングにより、正しいアミノ酸配列を持つタンパク質が合成される。スプライシングはゲノム配列によって厳格に規定されているが、遺伝多型によってスプライシングに異常が生じると、遺伝性疾患やがんを引き起こす原因となり得る。
[10] IL6(インターロイキン6)
免疫系で炎症反応の調節、免疫応答、細胞の成長と分化に関与する多機能なサイトカインの一種。主にマクロファージやT細胞などの免疫細胞から分泌され、感染や組織損傷に応答して迅速に生成され、炎症性サイトカインとして急性炎症反応を促進する役割を担う。
[11] Regnase-1
RNA結合タンパク質であり、免疫応答や炎症反応の調節において重要な役割を果たす。特に、炎症性サイトカインのmRNAの3’非翻訳領域に結合し、これを分解することで遺伝子発現を抑制し、過剰な炎症反応を防ぐ。IL6やTNF-αといったサイトカインのmRNAを標的とし、自己免疫疾患や炎症性疾患の発症リスクにも関連している。本研究では、3’非翻訳領域における遺伝多型を通じて、Regnase-1がIL6を制御するメカニズムとその臨床的影響を明らかにした。
形質とは個人の持つ性質のことをいい、病気の有無や身長などが含まれる。この中でも特に身長や白血球の数などは連続値であるため、量的形質と呼ばれる。本研究ではこの量的形質に着目して研究を行った。
[2] 遺伝多型
ゲノムは個人間でわずかな違いがある。この違いは一塩基の置換(一塩基多型)から、数十塩基までの挿入や欠失(挿入多型、欠失多型)、さらに大きな挿入や欠失(構造多型)などを含む。これらの遺伝多型の有無とヒトの形質を結び付けるのがGWAS([4]参照)である。
[3] 全ゲノムシークエンス
ヒト個体の全ての遺伝情報(ゲノム)を網羅的に解読する技術。ゲノムは約30億対のDNA塩基対から構成されており、この全体を読み取ることで、個体の遺伝的構成を詳細に把握できる。全ゲノムシークエンスはリファレンスパネル([7]参照)の作成に重要な役割を担う。
[4] ゲノムワイド関連解析(GWAS)
ヒトの形質(例:身長、病気のリスクなど)を目的変数とし、数百万に及ぶ遺伝多型を説明変数として統計学的検定を行い、ヒトの形質と有意に関連する遺伝多型を特定する手法。GWASはGenome-Wide Association Studyの略。
[5] 遺伝的インピュテーション
ゲノム解析において実際に測定されていない遺伝多型の情報を補完する手法。既知の遺伝情報(参照データ)を基に、欠損している部分の遺伝多型を推定し、データセットに含まれる遺伝多型数を増加させることで解析の精度を高める。
[6] 統計学的ファインマッピング
GWASで特定された関連領域の中から、原因となる可能性の高い遺伝多型を絞り込むための解析手法。GWASでは、広範なゲノム領域が検出されるが、その中には多くの遺伝多型が含まれている。統計学的ファインマッピングでは研究集団の連鎖不均衡(Linkage Disequilibrium:LD)情報を活用し、これらの遺伝多型の中から原因遺伝多型を特定する精度を向上させる。従来は計算量が多く、10万人を超える被験者や多数の遺伝領域に対しては実施が困難であったが、高効率なアルゴリズムの開発により可能となっている。
[7] リファレンスパネル
遺伝的インピュテーションにおける基準データであり、既知の遺伝多型情報を集めたデータセットのこと。全ゲノムシークエンスに基づいて収集された日本人集団の遺伝情報が多く含まれており、これを使用することで、実際に測定されていない遺伝多型を高精度で推定することができる。特に、対象集団の遺伝的背景と一致するリファレンスパネルを用いることで、インピュテーションの精度が向上する。
[8] 非コーディング多型
DNA配列の中でタンパク質の構造に直接関与しない領域に生じる遺伝多型。これらの非コーディング領域には、プロモーターやエンハンサー、イントロン、5’非翻訳領域、3’非翻訳領域などが含まれる。非コーディング多型は、タンパク質の構造には直接影響を与えないが、遺伝子の発現レベル、スプライシングの効率、転写因子の結合、mRNAの安定性や翻訳効率に影響を与え、結果として間接的に細胞の機能やタンパク質の産生に影響を与える可能性がある。
[9] スプライシング
前駆体mRNA(pre-mRNA)から不要な部分(イントロン)を除去し、必要な部分(エクソン)をつなぎ合わせて成熟したmRNAを形成する過程。このスプライシングにより、正しいアミノ酸配列を持つタンパク質が合成される。スプライシングはゲノム配列によって厳格に規定されているが、遺伝多型によってスプライシングに異常が生じると、遺伝性疾患やがんを引き起こす原因となり得る。
[10] IL6(インターロイキン6)
免疫系で炎症反応の調節、免疫応答、細胞の成長と分化に関与する多機能なサイトカインの一種。主にマクロファージやT細胞などの免疫細胞から分泌され、感染や組織損傷に応答して迅速に生成され、炎症性サイトカインとして急性炎症反応を促進する役割を担う。
[11] Regnase-1
RNA結合タンパク質であり、免疫応答や炎症反応の調節において重要な役割を果たす。特に、炎症性サイトカインのmRNAの3’非翻訳領域に結合し、これを分解することで遺伝子発現を抑制し、過剰な炎症反応を防ぐ。IL6やTNF-αといったサイトカインのmRNAを標的とし、自己免疫疾患や炎症性疾患の発症リスクにも関連している。本研究では、3’非翻訳領域における遺伝多型を通じて、Regnase-1がIL6を制御するメカニズムとその臨床的影響を明らかにした。

